CRT数据库是什么?它有哪些主要功能和用途?

CRA数据库,即Genome Sequence Archive(基因组序列归档库),是由中科院北京基因组研究所生命与健康大数据中心开发的原始组学数据归档库,该数据库在功能上等同于NCBI的SRA(Sequence Read Archive)、EBI的ENA(European Nucleotide Archive)和DDBJ的DRA(DNA Data Bank of Japan Archive),并且其数据模型和数据格式遵照INSDC标准。
CRA数据库的功能与特点
1、数据存储与访问:
CRA数据库用于存储二代测序的原始数据以及信息和元数据,这些数据通常以.sra文件格式保存,需要通过特定工具进行解压才能转换为可处理的fastq格式。
用户可以通过多种方式下载数据,包括直接使用wget命令从ftp服务器下载,或者使用专用工具如EdgeTurbo来提高下载速度和效率。
2、数据检索与下载:
用户需要先获取论文中的数据项目号(如CRA000167),然后在Genome Sequence Archive网站上进行搜索并下载对应的数据。
对于大量数据的下载,可以使用递归下载参数(如-r、-np等)来确保只下载所需目录及其子目录下的文件,同时避免下载无关内容。
3、数据验证:
下载完成后,建议使用md5sum等工具对下载的数据进行验证,以确保数据的完整性和准确性。
CRA数据库的使用案例
以下是一个具体的使用案例,展示了如何使用CRA数据库下载拟南芥的基因组序列和注释文件:
假设我们需要下载拟南芥的基因组序列和注释文件,首先我们需要找到对应的GEO号或SRR号,然后按照以下步骤操作:
1、查找GEO号或SRR号:
在论文中找到相关的GEO号或SRR号,GSEXXXXXX”。
2、访问NCBI网站:
打开NCBI官网(https://www.ncbi.nlm.nih.gov/),选择“GEO datasets”并输入GEO号进行搜索。
3、下载数据:
在搜索结果中找到所需的测序数据,点击进入后找到SRR号和下载链接。
使用sra-toolkit软件包中的prefetch命令下载数据,prefetch SRRXXXXXXXX。
4、解压缩数据:
使用fastq-dump工具解压.sra文件,得到双端测序的fastq文件:fastq-dump --split-files SRRXXXXXXXX.sra。
5、下载基因组序列与注释信息:
根据实验材料物种,下载相应的基因组序列与注释文件,对于拟南芥,可以下载TAIR10 genome release的所有基因注释文件和全基因组序列:
wget https://www.arabidopsis.org/Download_Files/Genes/GTF_genes_transposons.gff3.May2022.gff.gz
wget https://www.arabidopsis.org/Download_Files/Genome/tairan10_chr_all.fa.10.gz CRA数据库的优势与挑战
优势:
标准化:CRA数据库遵循INSDC标准,确保了数据的规范性和一致性。
高效性:通过专用工具如EdgeTurbo,用户可以快速高效地下载所需数据。
易用性:提供了详细的帮助文档和使用指南,方便用户上手和使用。
挑战:
数据量大:对于大规模数据集的下载和处理,可能需要较长时间和较多计算资源。
技术门槛:虽然提供了详细的使用指南,但对于初学者来说,仍然存在一定的技术门槛。
CRA数据库作为一个功能强大的基因组序列归档库,为科研人员提供了丰富的原始组学数据资源,通过合理的使用方法和工具,用户可以高效地获取和处理所需数据,从而推动科学研究的进展,面对海量的数据和复杂的技术要求,用户也需要具备一定的生物信息学知识和技能。



热门文章
-
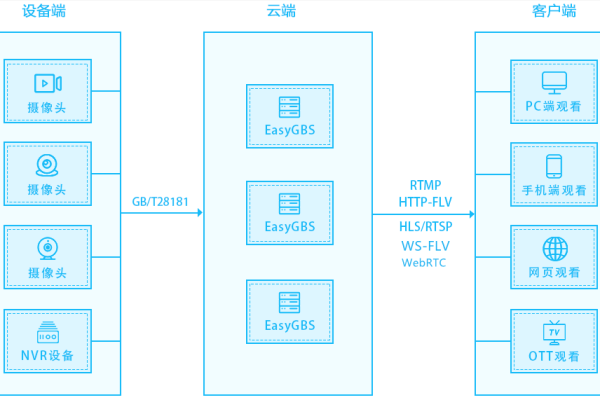
云服务器在搭建实时报警平台中扮演什么角色?
2024-10-04 -

MySQL存储过程的高效使用与编写指南,如何优化C语言中的MySQL存储过程?
2025-03-08 -

为什么服务器在技术世界中如此受到青睐?
2024-10-08 -
配件网站模板_网站模板设置
2024-06-23 -

微信中拉黑某人后,其头像显示状态会有什么变化?
2024-11-12 -

如何检查网络连接状态以确保设备已连接到互联网?
2025-03-01 -

如何轻松租用云服务器,关键步骤和技巧指南
2024-09-22 -
QQ资料卡为何显示为空?原因何在?
2024-11-11